Systemic amyloidosis results from extracellular deposition of fibrillar material derived from aggregation of precursor proteins into insoluble beta-pleated sheets. The most frequently recognized types are due to prolonged inflammation, deposition of immunoglobulin light chains, and accumulation of transthyretin, a tetrameric protein synthesized in the liver. Transthyretin amyloidosis can occur owing to wild-type or autosomal-dominant mutant transthyretin and predominantly affects the heart and peripheral nervous system. We present here 2 cousins with rapidly progressive cardiomyopathy secondary to TTR p.Val40Ile, a rare variant with high penetrance.1 Before genetic testing, each was thought to have an alternative diagnosis.
In the first case, a 59-year-old Caucasian man with hypertension, coronary disease, and carpal tunnel syndrome presented with subacute dyspnea and mildly elevated troponin. Coronary angiography showed no obstructive disease; transthoracic echocardiogram showed severe left ventricular hypertrophy with ejection fraction 55% and moderate right ventricular dysfunction. Cardiac magnetic resonance imaging (MRI) demonstrated septal-predominant left ventricular hypertrophy with subendocardial late gadolinium enhancement in the left ventricle, septum, and right ventricular free wall. Serum amyloid A, free light chains, and urine/serum protein electrophoresis were unremarkable.
Endomyocardial biopsy was consistent with wild-type transthyretin amyloidosis by immunohistochemistry and liquid chromatography/mass spectrometry. However, he was referred for clinical trial enrollment, where genetic testing identified the mutation TTR p.Val40Ile. He underwent heart transplantation less than 2 years after his original diagnosis.
In the second case, a 58-year-old Caucasian man with hypertension and hyperlipidemia presented with dyspnea and was diagnosed with hypertrophic cardiomyopathy. Cardiac MRI demonstrated septal-predominant left ventricular hypertrophy with transmural and epicardial foci of late gadolinium enhancement. Ten years later, genetic testing was negative for hypertrophic cardiomyopathy-linked genes but heterozygous for TTR p.Val40Ile.
Transthoracic echocardiogram showed severe concentric left ventricular hypertrophy with ejection fraction of 20%-25%, right ventricular dysfunction, and thickened mitral leaflets with mild–moderate regurgitation. Endomyocardial biopsy confirmed amyloidosis; laboratory tests were without evidence of monoclonal gammopathy, and serum amyloid A levels were normal. He rapidly deteriorated and required heart transplantation less than 1 year later.
Amyloid diagnosis has historically relied on histologic demonstration of amyloid deposits in biopsy specimens, which reveal characteristic green birefringence under cross-polarized light after Congo red staining.2 It is vitally important to distinguish amyloid subtypes, given differences in management and need for family screening. Immunohistochemisty, although widely available, cannot identify the fibril type in 20%-25% of cases.3 Mass spectrometry has emerged as the gold standard for fibril typing, using computer algorithms to match peptides to a reference database. However, in one recent study of 56 genotype-positive transthyretin amyloidosis patients, liquid chromatography/mass spectrometry failed to identify pathogenic mutations in 9 individuals.4
Noninvasive cardiac imaging can assist in diagnosis and disease surveillance. Transthoracic echocardiography may demonstrate biventricular hypertrophy, valvular thickening, biatrial dilatation, and impaired strain.2 Bone tracer scintigraphy (eg, Tc99-pyrophosphate) is reported to be >99% sensitive and 86% specific for transthyretin amyloidosis,5 but is semiquantitative and does not evaluate cardiac structure or function. Cardiac MRI is emerging as an important diagnostic tool. In 263 patients with scintigraphic evidence of cardiac transthyretin amyloidosis, 79% had asymmetric septal hypertrophy, 71% transmural late gadolinium enhancement, and 96% right ventricular late gadolinium enhancement. Additionally, extracellular volume paralleled scintigraphic cardiac uptake and predicted mortality.6
Transthyretin amyloidosis is increasingly recognized in patients with heart failure, aortic stenosis, or a hypertrophic cardiomyopathy-like phenotype.7 Peripheral neuropathy, carpal tunnel syndrome, tendon tears, and spinal stenosis may also provide diagnostic clues to transthyretin amyloidosis.8 Although transthyretin amyloidosis treatment is currently limited, novel transthyretin-stabilizing and gene-silencing therapies hold promise for organ recovery and increased survival.2 A high index of suspicion and appropriate utilization of genetic testing can ensure timely and accurate diagnosis for patients and their family members.
-Jessica R. Golbus, MD, Joanna M. Wells, BS, Michael G. Dickinson, MD, Scott L. Hummel, MD, MS
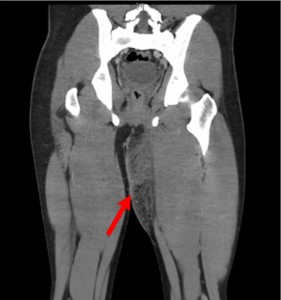 Significant cardiac signs and symptoms developed in a young patient undergoing treatment for a severe infection. The 23-year-old man presented with a left groin abscess and a 5-day history of fever. Computed tomography (CT) showed findings consistent with fasciitis (Figure 1). He was initially treated with intravenous vancomycin, ampicillin-sulbactam, and clindamycin. On hospital day 2, he developed severe chest pain, dyspnea, and nonsustained ventricular tachycardia.
Significant cardiac signs and symptoms developed in a young patient undergoing treatment for a severe infection. The 23-year-old man presented with a left groin abscess and a 5-day history of fever. Computed tomography (CT) showed findings consistent with fasciitis (Figure 1). He was initially treated with intravenous vancomycin, ampicillin-sulbactam, and clindamycin. On hospital day 2, he developed severe chest pain, dyspnea, and nonsustained ventricular tachycardia.













